
研究背景
催化反应网络(Catalytic Reaction Networks, CRNs)是连接宏观反应动力学与原子尺度催化机理的重要理论框架。在多相催化过程中,吸附、表面反应和产物脱附等基本步骤相互耦合,构成庞大而复杂的反应网络。对于 CO2加氢、费托合成等涉及大量中间体和基元反应的体系,若完全依赖传统 DFT方法逐一构建和求解反应网络,计算成本极其高昂,往往难以承受。
过去,研究人员通常通过预设主反应路径或借助 Brønsted-Evans-Polanyi关系进行近似,从而降低计算量。但这类方法常常牺牲准确性,难以完整刻画反应路径之间的竞争关系。近年来,机器学习原子间势(MLIPs)为催化研究提速提供了新思路,但预训练模型在不同泛函、不同计算参数乃至不同催化体系下往往存在泛化能力不足的问题。
因此,如何在计算效率与理论精度之间取得平衡,成为复杂催化反应网络自动化探索中的关键挑战。
研究方法
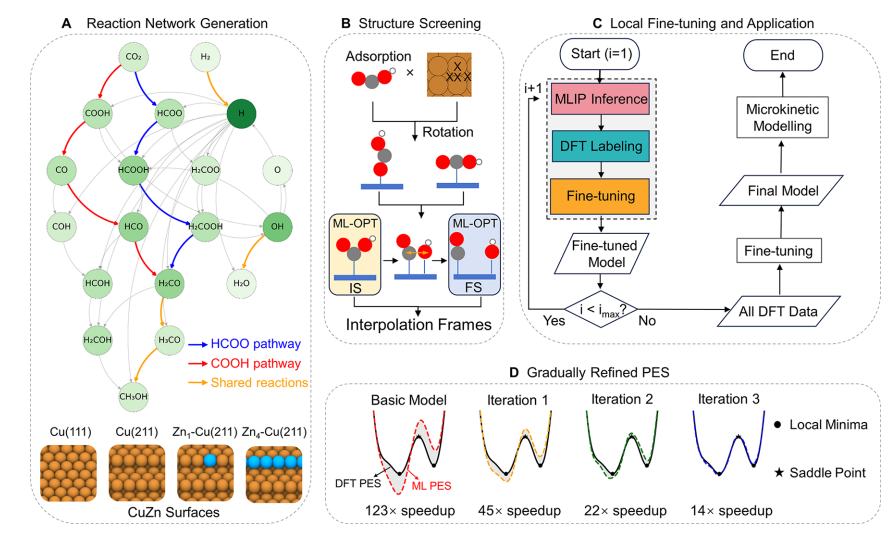
针对上述问题,南开大学刘锦程团队提出了 LFT-CRN 框架。该方法将预训练通用机器学习原子间势与局域微调主动学习策略相结合,通过“机器学习推断—DFT标注—模型微调”的迭代流程,不断修正势能面,使模型在局部化学空间中快速逼近 DFT精度。具体而言,研究首先针对 Cu(111)、Cu(211)、Zn1-Cu(211) 和 Zn4-Cu(211) 四类代表性 CuZn表面建立模型,围绕 CO2制甲醇反应构建包含 19个中间体和 23个基元反应的催化反应网络。随后,研究团队利用预训练模型对吸附构型、初态/终态结构和 NEB路径进行快速筛选与优化,再通过 DFT对关键结构进行标注,并将这些新数据加入训练集进行局域微调。在此基础上,研究进一步构建统一模型,用于振动频率与零点能(ZPE)预测,并结合 CatMAP 进行微观动力学模拟,从而系统分析不同 CuZn表面的甲醇合成活性。
研究结果
研究结果表明,LFT-CRN在多个关键任务中均实现了显著提速与高精度兼顾。
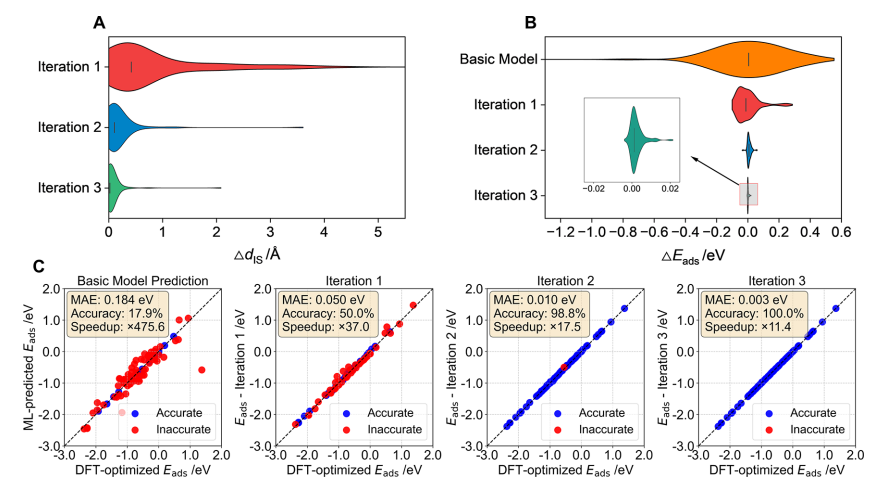
在吸附结构优化方面,经过三轮局域微调后,吸附能预测的平均绝对误差(MAE)从初始模型的 0.184 eV大幅下降至 0.003 eV,并保持约 11.4倍 的计算加速比。
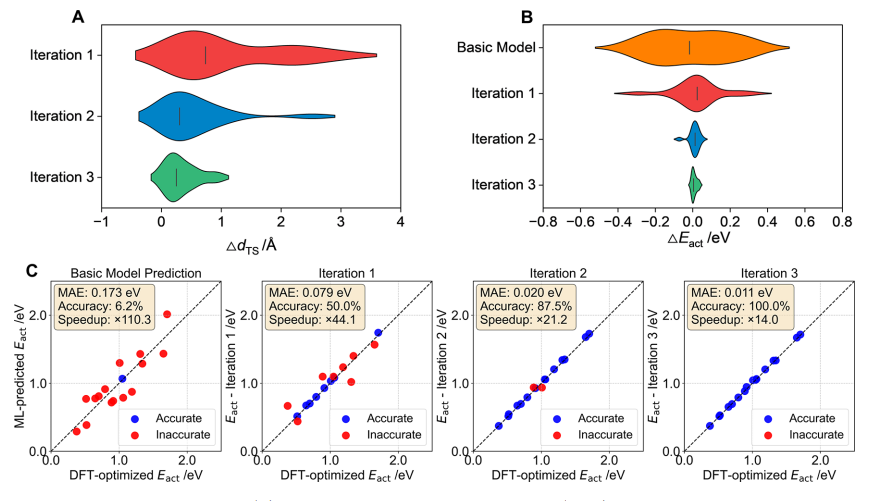
在过渡态搜索方面,LFT-CRN将活化能预测误差从 0.173 eV降低至 0.011 eV,预测准确率提升至 100%,同时相较传统 DFT-NEB流程实现约 14.0倍的加速。
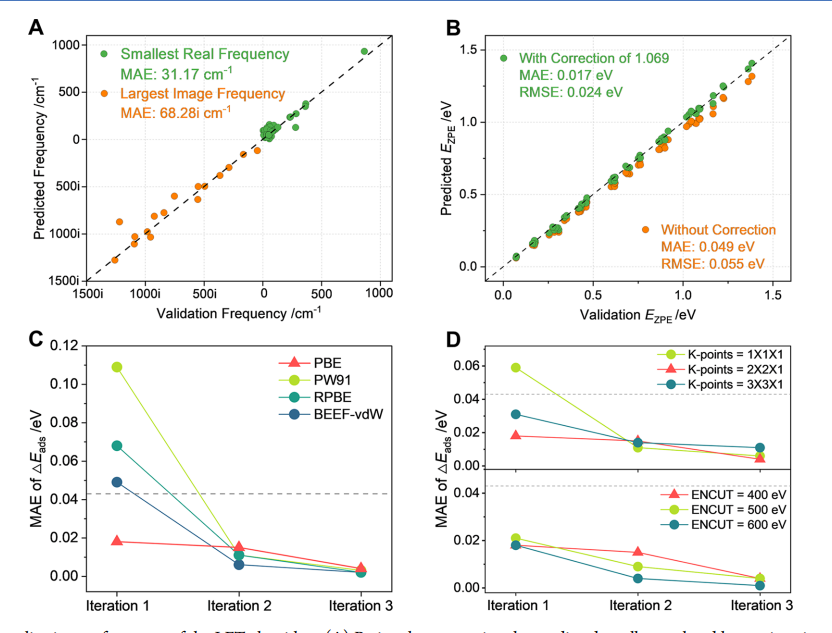
在振动频率分析方面,统一微调模型对最小实频和最大虚频均表现出良好预测能力,相对常规 DFT频率计算实现约 195.5倍提速。经缩放修正后,零点能预测误差可降至 0.017 eV,达到化学精度要求。
从整体工作流看,针对四类 CuZn表面的甲醇合成反应网络,传统 DFT流程在单 CPU节点上约需 50天,而 LFT-CRN 仅需 2.3-3.5天 即可完成,整体加速达到 14.2-21.5倍。
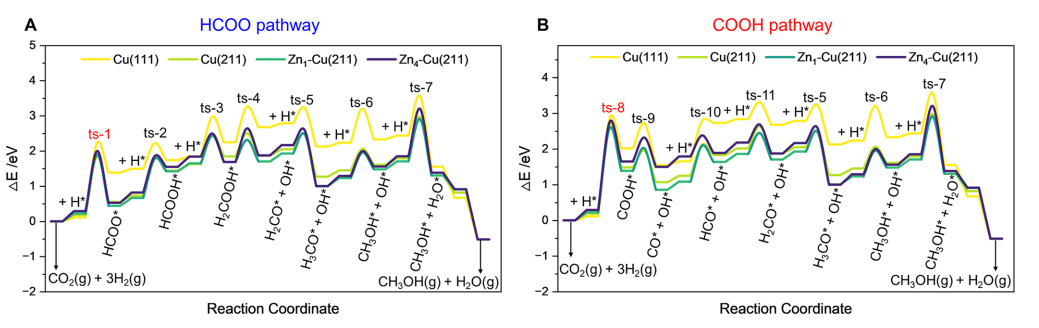
在催化机理方面,研究显示:Cu(111)表面的整体能量轮廓最高,对甲醇合成贡献最弱;Cu(211)与 Zn1-Cu(211)均表现出较高活性,其中后者对多数中间体具有更强吸附能力;Zn4-Cu(211)因 Zn覆盖过高,整体能量景观升高,不利于甲醇生成。
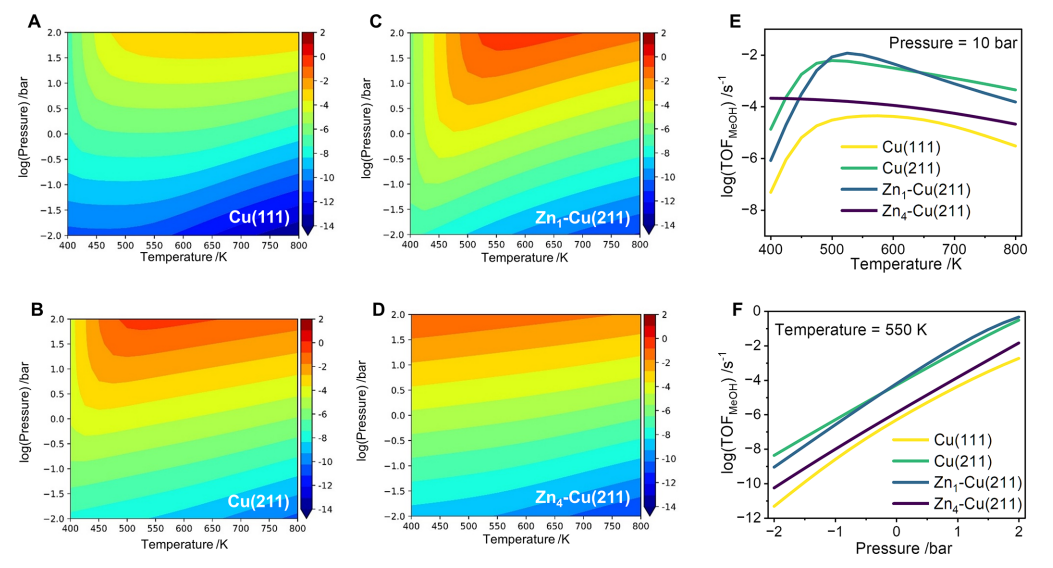
微观动力学模拟进一步表明,在工业相关条件下(如 550 K、10 bar),Zn1-Cu(211)的甲醇生成周转频率约为 Cu(211)的两倍,是四种表面中活性最高者。这说明,低配位 Cu位点与适度 Zn掺杂所形成的 Cu-Zn协同作用最有利于提升 CO2加氢制甲醇性能,而过量 Zn会削弱催化活性。
期刊:ACS Catalysis
题目:Accelerating Catalytic Reaction Network Exploration via Local Fine-tuning with Universal Machine Learning Interatomic Potentials
作者:Pengfei Hou, Jingshan Luo, Jin-Cheng Liu*
接受日期:17 March 2026
原文链接:https://doi.org/10.1021/acscatal.5c08361

