异核双金属位点催化剂具(DACs)有独特的配位结构,能够精确调控金属自旋态,为高效活化过一硫酸盐(PMS)提供了巨大潜力。尤其重要的是,该类催化剂可实现高价钴-氧物种(Coᴵⱽ=O)的高效生成。然而,具有轴向配位双原子结构的DACs的识别及其机制功能仍不明确。
2025年11月8日,南开大学卜显和院士、沈铸睿、李娜团队合作在Journal of the American Chemical Society期刊发表题为“Axial Co-O-Cu Pairs Enable Superexchange Interaction for Efficient Coᴵⱽ=O Formation toward Water Purification”的研究论文,南开大学陈方园为论文第一作者,沈铸睿、李娜为论文共同通讯作者。

第一作者:陈方园
通讯作者:沈铸睿、李娜
通讯单位:南开大学
论文DOI:10.1021/jacs.5c16247
该研究展示了一种锚定在氮化碳(CoCu-CN)中的具有轴向Co–O–Cu对(CoN₄-O–CuN₄)的DAC。该独特构型显著稳定了Co的中自旋态,从而增强了PMS活化以高效生成Coᴵⱽ=O。结合实验与理论分析表明,通过Co 3d(dxz/dyz)轨道与O p(px/py)轨道的超交换介导了从Co到Cu的电子转移。这种强烈的Co–O杂化将d带中心上移至–0.41 eV,促进了PMS的吸附。此外,Co和Cu中d轨道的轨道极化优化促进了PMS中O–O键和O–H键的同时断裂,从而实现了直接的Coᴵⱽ=O形成。因此,该催化剂设计实现了优异的降解性能,包括磺胺异噁唑降解速率达到1.98 min⁻¹,分别是Co–CN和Cu–CN的5.8倍和19.8倍。该工作为设计同时具备自旋活性和持久稳定性的PMS活化催化剂确立了新范式。
面对水污染中有机污染物(如抗生素)的持续威胁,基于过氧化物的高级氧化工艺(AOPs)因其强氧化能力已成为污染物降解的关键技术。其中,与基于自由基的途径相比,高价金属-氧物种提供了更优异的选择性降解能力。然而,从过一硫酸盐(PMS)等过氧化物中稳定生成此类物种受到对于具有高d电子数金属(d>4,例如Coᴵⱽ)的“氧墙规则”的限制。因此,开发能够打破“氧墙规则”以实现靶向且高效生成高价金属-氧物种的催化剂,是提高PMS-AOPs选择性和效率的迫切挑战。精确调控催化剂的电子结构至关重要,其中金属中心的自旋态是控制氧化反应中O–O键和O–H键裂解路径的关键调控参数。
异核双原子催化剂(DACs)为在PMS活化过程中调控自旋态提供了一个独特的平台,从而克服“氧墙规则”并实现Coᴵⱽ=O的靶向生成。具体而言,具有轴向桥连配位的双金属中心(M−L−M:金属−配体−金属,其中M为磁性阳离子)通过超交换相互作用促进自旋调控。然而,该效应的有效建立取决于满足两个关键条件:原子级精确的M–L–M键角以及金属对之间的电子互补性。接近线性或垂直的几何构型确保了超交换相互作用,而供体-受体组合有助于抑制自旋不稳定性。尽管该机制在结构明确的晶体材料(如金属氧化物/氟化物)中已得到充分证实,但像氮化碳这样的非晶材料因其独特的优势而备受关注。这些非晶载体具有丰富的表面悬键以增强金属锚定、可调的局域电子结构以优化反应路径,以及高比表面积以暴露更多活性位点。然而,非晶载体固有的结构无序性极大地降低了形成所需的有效M₁-L–M₂配位的概率。此外,非晶载体中的结构无序阻碍了自旋耦合。因此,在非晶载体上对轴向配位双原子位点的自旋态进行原子尺度调控仍需深入研究。这是开发能够通过自旋态工程克服“氧墙规则”的新型、更高效催化剂的关键。
在此,该研究展示了一种氮化碳负载的Co和Cu双原子催化剂(CoCu-CN),其具有原子级精确的轴向配位(CoN₄-O–CuN₄)。接近线性的Co–O–Cu对促进了金属位点间的超交换相互作用。在此机制中,闭壳层Cu⁺位点将Co³⁺位点稳定在中自旋态。因此,促进了一种非经典的PMS活化路径,其中分子内氢键与增强的Co–O₁键协同作用,裂解PMS的O₁-O₂键和O₁-H键,从而快速生成Coᴵⱽ=O活性物种。优化的电子构型带来了卓越的催化性能:磺胺异噁唑降解速率达到1.98 min⁻¹,分别是参比材料的5.82倍和19.8倍。此外,在连续运行6小时后,CoCu–CN对罗丹明B仍保持96%的降解效率,显示出优异的抗自旋弛豫能力和结构稳健性。该设计为开发集自旋活性与长期循环稳定性于一体的PMS活化催化剂确立了新范式。
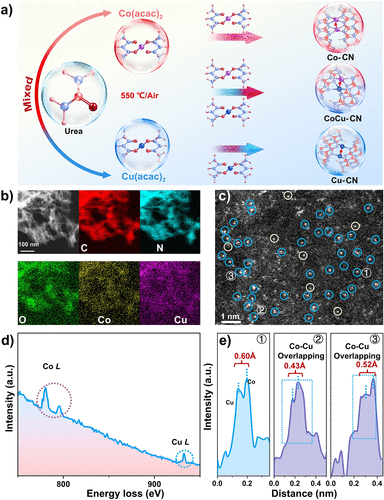
图1. 双原子催化剂的合成与原子尺度表征。(a) CoCu–CN、Co–CN和Cu–CN的合成路线示意图。(b) CoCu–CN的EDS映射图,显示C、N、O、Co和Cu的均匀分布。(c) CoCu–CN的AC-HAADF-STEM图像(双原子和单原子分别用蓝色和浅黄色标记)。(d) 从(c)中双原子位点获取的EELS谱。(e) 对应于(c)中标记位点1–3的强度分布曲线。
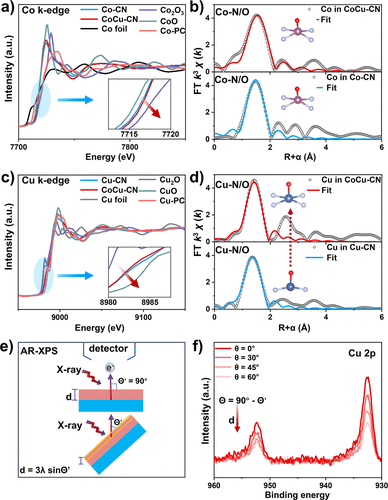
图2. (a, c) 不同样品中Co和Cu的K边XANES谱。插图:谱图的放大区域。(b, d) Co和Cu的EXAFS谱的傅里叶变换幅度。(e) AR-XPS中关于电子出射角的表面成分分布对XPS信号强度影响的示意图。(f) 在不同出射角下收集的CoCu–CN的Cu 2p XPS谱。
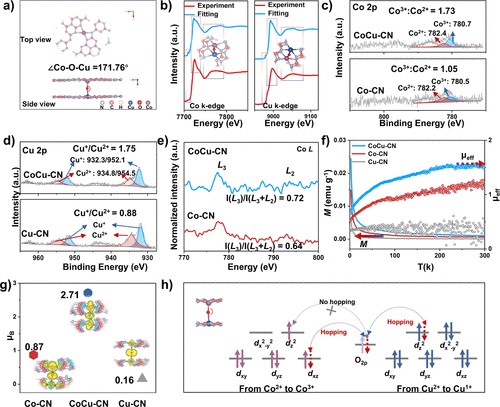
图3. (a) CoCu−CN的DFT优化结构示意图。(b) Co/Cu K边的实验XAFS谱及计算得到的XANES拟合谱。(c, d) 催化剂的Co 2p和Cu 2p XPS谱。(e) 催化剂的Co L边sXAS谱。(f) 催化剂的磁化强度随温度变化曲线及有效磁矩随温度变化曲线。(g) 不同催化结构的磁矩。(h) CoCu−CN结构中超交换模型的示意图。
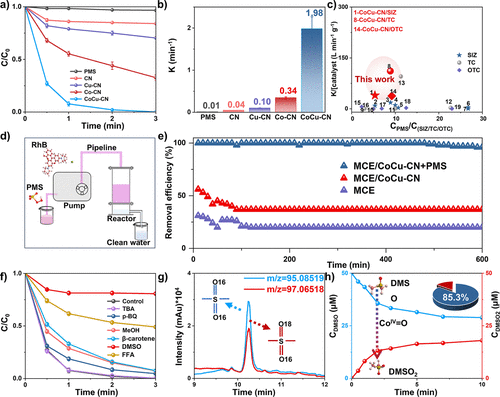
图4. (a, b) 不同催化剂的降解效率和降解反应动力学常数。(c) CoCu–CN与文献中报道的其他催化剂对SIZ、TC和OTC降解速率的比较。(d) 循环稳定性测试装置示意图;(e) 使用循环稳定性装置对RhB的连续去除效率;(f) CoCu–CN/PMS/SIZ体系中的猝灭实验。(g) CoCu–CN活化的PMS体系中DMSO的氧化和DMSO₂的形成。(h) CoCu–CN/PMS体系中DMSO氧化过程中生成的DMSO¹⁶O¹⁶和DMSO¹⁶O¹⁸的提取离子色谱图。反应条件:CSIZ = 5 mg/L, CPMS = 0.1 mM, 催化剂浓度 = 0.05 g/L, 温度 = 298 K, 初始pH = 7。
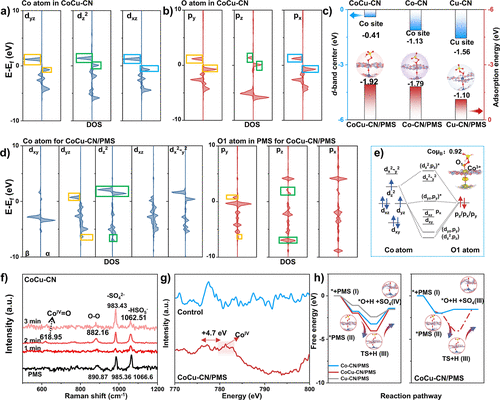
图5. (a, b) CoCu–CN催化结构中Co 3d轨道和O 2p轨道的PDOS。(c) 不同构型中金属位点的d带中心及相应的吸附能。(d) CoCu–CN/PMS吸附构型中Co 3d轨道和O1 2p轨道的PDOS。(e) CoCu–CN中Co 3d轨道与PMS中O 2p轨道之间电子耦合示意图。(f) CoCu–CN/PMS体系的Operando拉曼光谱。(g) CoCu–CN和CoCu–CN/PMS在Co L边的归一化sXAS谱。(h) 沿PMS活化路径形成Coᴵⱽ=O物种的自由能图。
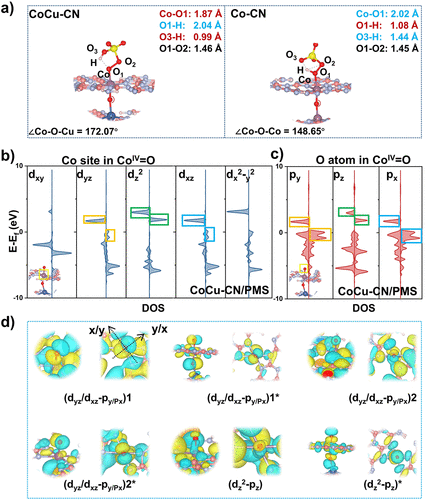
图6. (a) CoCu–CN/PMS和Co–CN/PMS体系中吸附构型的键长分析。(b, c) CoCu–CN中Coᴵⱽ=O物种的Co 3d和O 2p轨道的pDOS。(d) 轨道相互作用后的波函数空间分布。
总之,该研究展示了一种异核自旋解耦策略,用于在CoCu–CN中构建具有轴向Co–O–Cu对的双原子催化剂,克服了PMS活化生成高价Coᴵⱽ=O物种过程中“氧墙规则”和自旋弛豫的限制。由闭壳层Cu⁺介导、Co–O–Cu键角为171.76°的超交换相互作用持续地将Co³⁺稳定在中自旋态。同时,XPS和软XAS分析证实了Cu⁺物种的97%富集以及Co³⁺氧化态65%的增强。强烈的Co–O轨道杂化诱导CoCu–CN中Co的d带中心显著上移(0.72 eV至–0.41 eV)。理论和实验研究共同揭示,Co–O–Cu结构驱动了分子内氢键(O₃−H键:0.99 Å)的形成,从而实现了一种非经典的PMS裂解路径,将Coᴵⱽ=O形成能垒降低至0.36 eV。通过猝灭实验、¹⁸O同位素示踪、原位拉曼光谱和软XAS明确证实了Coᴵⱽ=O作为主要活性物种的主导地位。该设计实现了卓越的性能:SIZ降解速率达到1.98 min⁻¹(较Co-CN和Cu-CN高5.8-19.8倍),并且在连续运行10小时的膜反应器操作后仍保持>96%的污染物去除效率。该工作提出了一种成功将自旋态活性与有前景的工业级稳定性相结合的催化体系,为在复杂废水处理环境中实施非自由基氧化过程提供了可行策略。
期刊:Journal of the American Chemical Society
题目:Axial Co−O−Cu Pairs Enable Superexchange Interaction for Efficient CoIV=O Formation toward Water Purification
作者:Fangyuan ChenRan ZhaoHexiang ZhaoQian LiuShiyi ZhaoNa Li*Zhurui Shen*Xian-He Bu
接受日期: 8 November 2025
原文链接: https://doi.org/10.1021/jacs.5c16247

