
南开大学刘锦程团队J. Am. Chem. Soc.: M-N-C单原子催化剂中电势驱动吸附能的反转
--- 材料科学与工程学院-中文 ---
--- 材料科学与工程学院-中文 ---
来源:科研任我行

原子分散的金属原子锚定在氮掺杂石墨烯材料(N-C)中的多相单原子催化剂(SACs)因其在电化学反应中的应用而备受关注,如氧还原反应(ORR)和二氧化碳还原反应(CO2RR)。在这些催化剂中,金属原子通常与吡啶氮原子形成四个配位键。这些材料的催化效率与其电子结构密切相关,电子结构因金属类型、配位原子和周围环境而异。特别是,电子结构对于理解金属单原子催化剂(SACs)的催化机理至关重要,尤其是在电化学条件下。
在此研究中,作者深入研究了电化学电势对N-C衬底上SACs中“前线轨道”的细微调制。研究观察到费米能级的变化和d轨道占据随电化学电势的变化而变化,强调了金属的离散原子轨道和基于N-C环境的连续能带之间的协同作用。使用O2和CO2作为模型吸附物,研究强调了这些变化对吸附能的直接影响,揭示了在负电化学电势下Co/N-C SAC上吸附能的有趣反转。这种发现归因于dxz和dz2轨道的作用,它们对稳定O2的π*轨道至关重要。通过这一探索,这项研究工作为SACs中电子结构和吸附行为之间的相互作用提供了见解,为增强电化学过程中催化剂的设计策略铺平了道路。
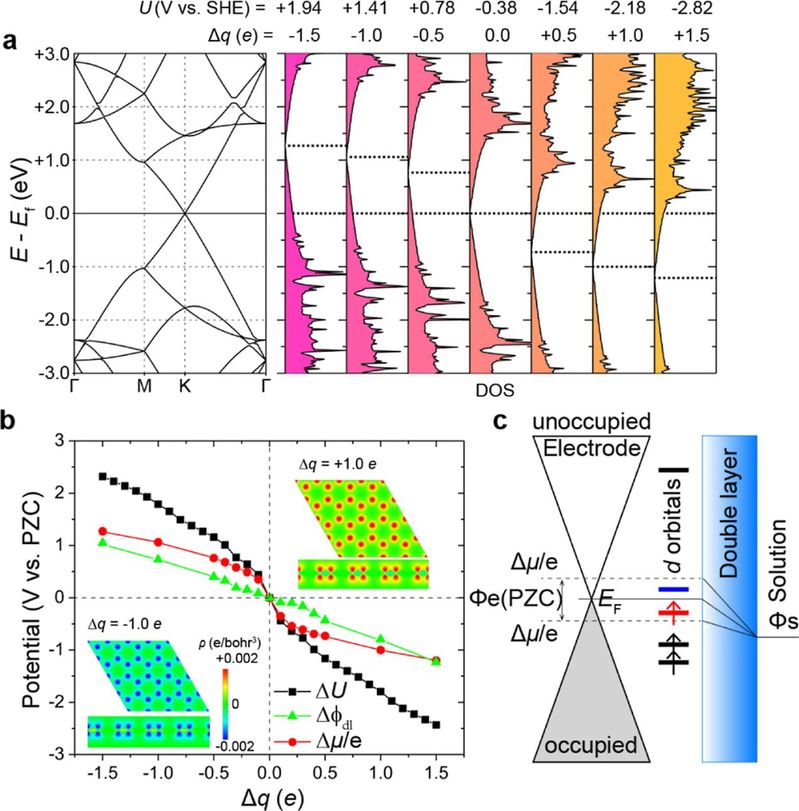
图1 石墨烯的电子结构
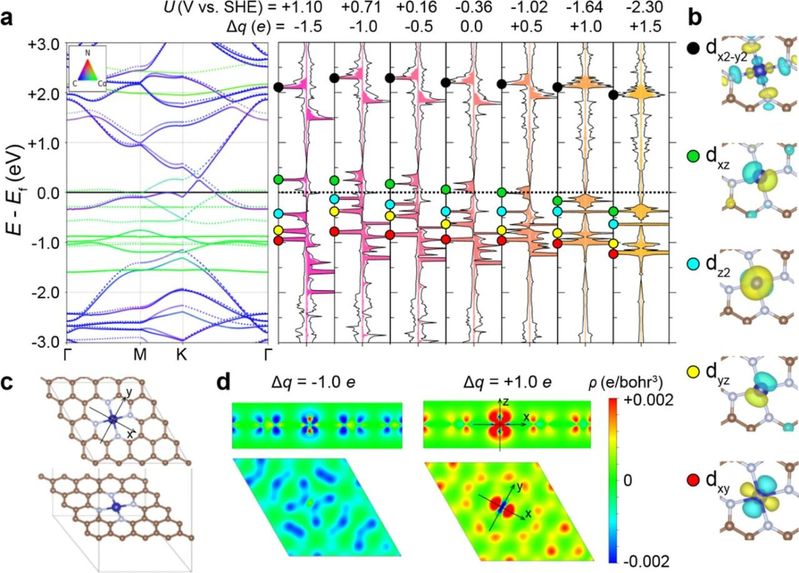
图2 Co/N-C的电子结构
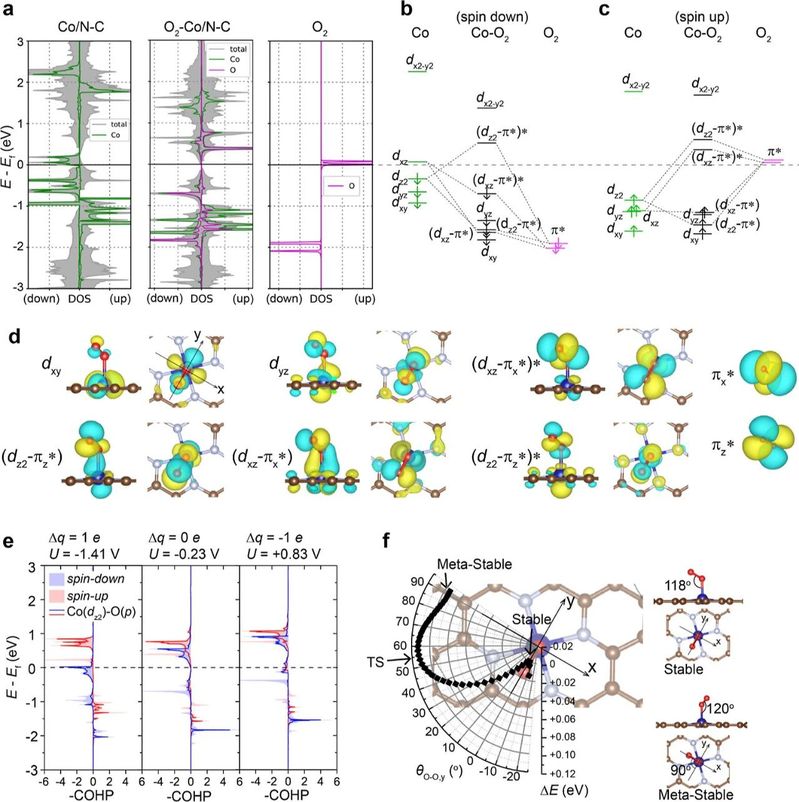
图3 Co/N-C上的O2吸附
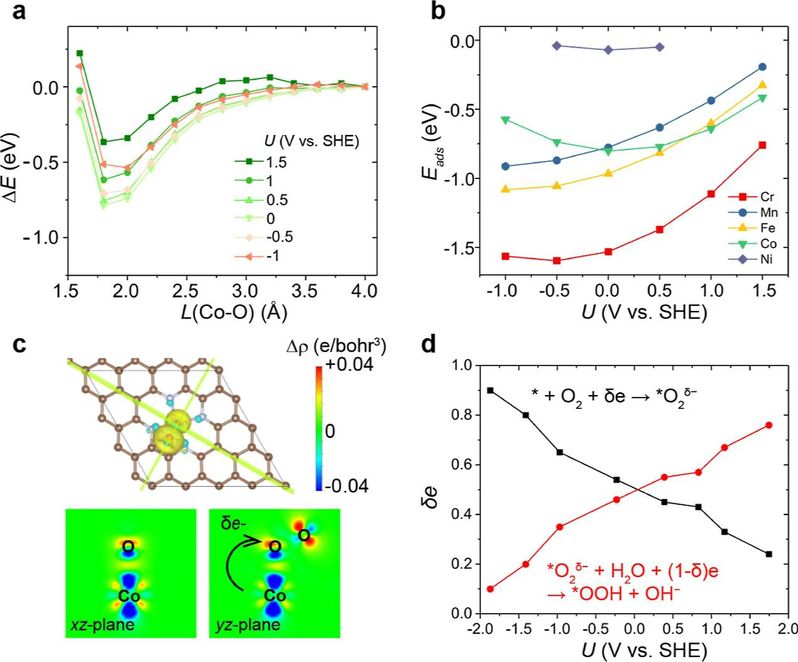
图4 不同电势下M/N-C上的O2吸附
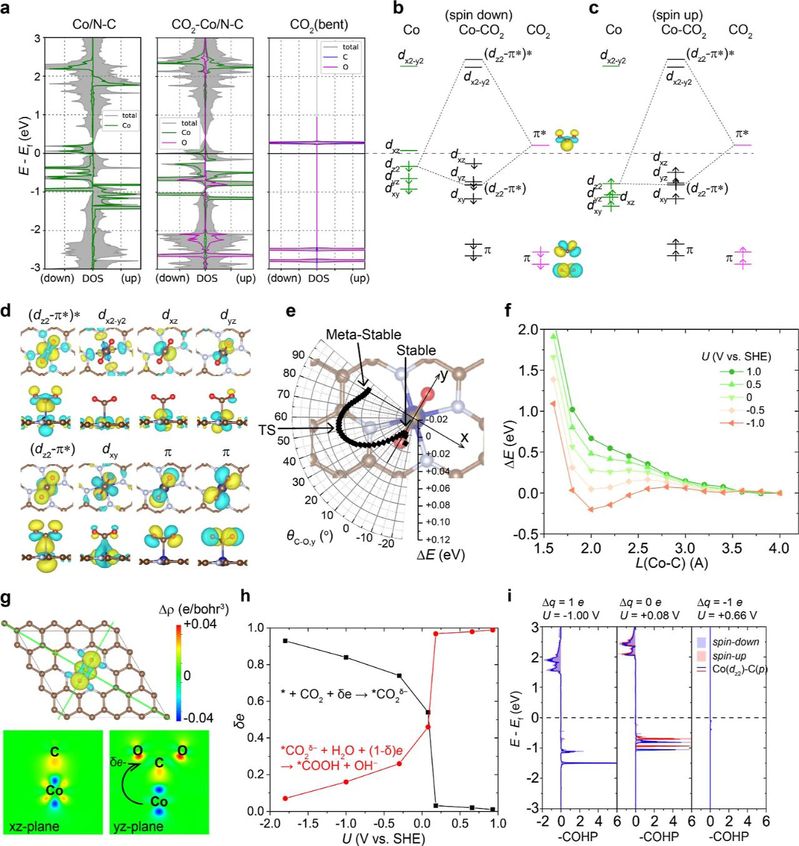
图5 Co/N-C上的CO2吸附